
赵文芝,石汉平(首都医科大学附属北京世纪坛医院胃肠外科/临床营养科/肿瘤代谢与营养北京市国际科技合作基地/首都医科大学肿瘤学系,北京 100038)
【正文】
维生素C,又称抗坏血酸,是由葡萄糖转化而来的六碳酮内酯,在新鲜的蔬菜水果中含量丰富。它具有抗氧化,清除自由基,促进类固醇代谢,改善钙、铁和叶酸的利用,促进羟化反应等重要的生理作用【1】。维生素C在细胞内/外环境中均易被氧化,细胞外环境中的自由基或活性氧家族(reactive oxygen species,ROS)均可氧化维生素C,进而形成脱氢抗坏血酸(dehydroascorbic acid,DHA)等氧化形式,此时这些变化是可逆的,DHA仍具有维生素C的活性;但DHA发生进一步氧化反应后则失去维生素C活性,为不可逆反应【2,3】,详见图 1。
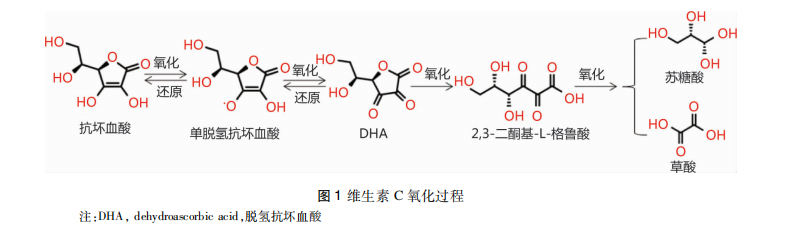
维生素C的还原特性使其表现出抗氧化、抗衰老、预防和治疗缺铁性贫血的生理作用。维生素C还是某些酶的辅助因子,如具有催化生物合成和基因调控作用的单加氧酶和双加氧酶家族,这些酶参与胶原、肉碱、儿茶酚胺类激素、酰胺化肽激素的合成,并能羟化转录因子,如低氧诱导因子-1α(hypoxia-inducible factor 1-α,HIF-1α)、甲基化的DNA、甲基化的组氨酸等【4,5】,因此维生素C还表现出促进胶原形成、类固醇代谢及防癌抗癌等作用。
将维生素C用于肿瘤治疗的研究始于20世纪70年代,1974 年Cameron C等【6】首先通过临床试验研究发现,在进展期肿瘤患者中经静脉给予大剂量维生素C(10g/d,持续10d治疗),后持续口服维生素C (10g/d)以维持血药浓度,可改善患者的症状和生存情况;此后扩大的临床试验进一步证实了大剂量维生素C补充应用于终末期肿瘤患者治疗时的作用,补充大剂量维生素C后受试者平均生存期延长了 3.2倍(对照组平均50d,补充组平均210d)【7,8】。 这个发现引起了人们的极大关注,维生素C在肿瘤患者中的应用及其抗肿瘤研究迅速兴起。但是,好景不长,1985年由美国Mayo诊所 Creagan ET【9】及 Moertel CG等【10】开展的两项设计合理的随机对照临床试验。
试验一:试验组60例,对照组63例,一般情况及疾病情况类似;
试验二:在100例进展期结直肠癌患者的进行双盲随机试验,试验组、对照组纳入试验前均未进行肿瘤化学治疗。
未能重现以上结果,即虽然肿瘤患者口服了同样剂量的维生素C(10g/d),其生存情况及症状并未获得改善。
◆试验一: 与对照组相比,试验组的症状、功能表现、食欲或体重无显著改善,两组中位生存时间均为 7 周,生存曲线重合;
◆试验二:与对照组相比,试验组在症状改善率(对照组:65%,实验组:64%)、生存期(中位生存期,对照组 4.1年,试验组 2.9年)方面无明显优势。
自此,维生素C被打入肿瘤治疗的“冷宫”。到了21世纪,关于维生素C的药代动力学研究发现,维生素C经口服和静脉两种方式进入人体后,血药浓度差别甚大,口服给药很难突破 200μM(平衡的膳食可使血浆维生素C达30~80μM),而经静脉给药血药浓度可达10μM以上【11,12】。此时, 人们回头审视Cameron ET 和Moertel CG等【11,13】两派的研究,推断大剂量维生素C肿瘤治疗效果的矛盾结论可能与其用药方式有关。于是,维生素C重新成为抗肿瘤研究的一个热点【14】。
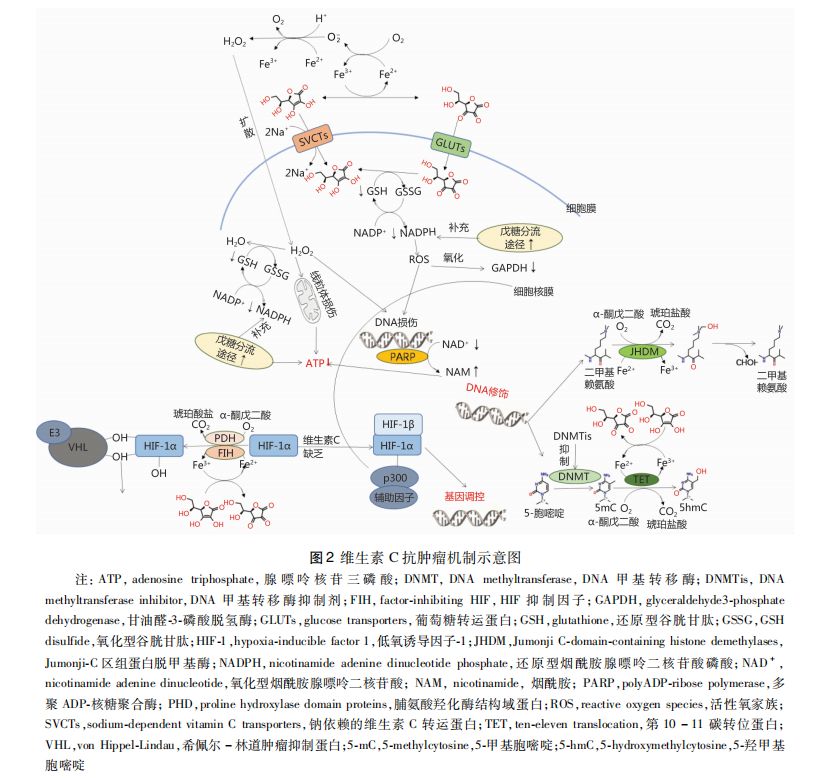
随着研究的深入,维生素C的抗肿瘤机制正在被一步步揭示,目前比较被认可的机制主要有3种, 即维生素C诱导产生过氧化氢(H₂O₂)及其导致的过氧化应激、维生素C介导的表观遗传学改变、维生素C调节缺氧诱导因子(hypoxia-inducible factor,HIF)活性。
1.维生素C诱导的过氧化应激
细胞外液中,维生素C的不同活性形式的相互转变过程催化生成 H₂O₂ ,体外试验中,药理学浓度(毫摩尔级)的维生素C可在培养基中自氧化生成H₂O₂【15】;而体内试验数据显示,细胞外液单脱氢抗坏血酸浓度达100nmol/ L以上时即可形成 H₂O₂【11】。穿过细胞膜进入细胞通过耗竭细胞能量,最终导致细胞死亡,该过程可能涉及:
① H₂O₂造成细胞内DNA链断裂,该损伤需由多聚ADP-核糖聚合酶(polyADP-ribose polymerase,PARP)修复,PARP修复DNA链的过程需要消耗氧化型烟酰胺腺嘌呤二核苷酸(nicotinamide adenine dinucleotide,NAD﹢ ) ,NAD﹢作为辅助因子被分解为烟酰胺,而同时NAD﹢作 为 甘 油 醛-3-磷 酸 脱 氢 酶 (glyceraldehyde3-phosphate dehydrogenase,GAPDH)的辅助因子,NAD﹢的耗竭会直接影响由 GAPDH催化的生成ATP的反应,导致细胞内ATP下降【16,17】;
②H₂O₂可能由谷胱甘肽过氧化物酶催化的氧化还原反应消除, 即在该酶的催化下还原型谷胱甘肽(glutathione,GSH)被H₂O₂氧化为氧化型(GSH disulfide,GSSG),细胞内 GSSG再次被还原为 GSH的过程消耗还原型烟酰胺腺嘌呤二核苷酸磷酸(nicotinamide adenine dinucleotide phosphate,NADPH ) ,而NADPH需要通过戊糖分流途径消耗更多的葡萄糖来进行补充,进而导致细胞内葡萄糖缺乏、能量产生不足【18,19】;
③H₂O₂可能导致或加重线粒体功能障碍,直接影响能量的生成过程【19,20】。
另一个方向的研究未关注H₂O₂的产生及作用,而专注于维生素C引起的细胞内的过量 ROS。肿瘤细胞表现出独特的瓦博格效应 (Warburg effect),即肿瘤细胞糖酵解反应活跃,葡萄糖摄取率高、代谢产物乳酸含量高、能量产生效率低,即使在氧气充足的情况下也不能改变此代谢模式【21,22】。维生素C的活性形式通过两种途径进入细胞,维生素C原型通过钠依赖的维生素C转运蛋白(sodium-dependent vitamin C transporters,SVCTs)以耗能的方式进细胞,DHA因其分子结构类似葡萄糖而通过葡萄糖转运蛋白(glucose transporters,GLUTs)以易化扩散的方式进入细胞,某些细胞摄取DHA的效率高于维生素C原型【23,24】。DHA进入细胞后被细胞内的GSH、硫氧还蛋白(thioredoxin)和 NADPH迅速还原为维生素C原型,导致细胞内还原物质耗竭、ROS水平升高,类似H₂O₂的影响,升高的 ROS会氧化GAPDH使其失活、激活PARP进而耗竭NAD﹢,最终导致细胞能量耗竭【25,27】。
虽然关于维生素C引起氧化应激损伤的主要机制仍存在分歧,即主要作用物质为H₂O₂还是 DHA,但关于大剂量维生素C杀伤肿瘤细胞的机制研究方面过氧化应激假说仍占主导地位【28】。
2.维生素C调节表观遗传学改变
维生素C通过作用于氧化还原活性铁(Fe²+/Fe³+)调节铁离子-α-酮戊二酸依赖的双氧合酶(Fe²+-and α-ketoglutarate-dependent dioxygenases,Fe²+/α-KGDDs),进而参与包括脯氨酸羟化酶、组蛋白脱甲基酶、 烷烃羟化酶(alkane hydroxylase,ALKB) 同系物和第10-11碳转位(ten-eleven translocation,TET)蛋白等在内的多种酶催化的多种生物过程【29-31】。 维生素C作为辅助因子增强 TET 蛋白的活性,导致DNA脱甲基化、羟甲基化作用增强,而这些变化最终的影响是:导致肿瘤细胞中肿瘤抑制基因的重新表达、促进干细胞分化、增强 DNA甲基转移酶抑制剂(DNA methyltransferase inhibitor,DNMTi)诱导的免疫信号【31-33】。DNA甲基化是肿瘤发生的早期事件,DNA超甲基化多见于抑癌基因、肿瘤发展基因的启动【34】;某些肿瘤,尤其是血液肿瘤常表现出异常的高甲基化[ DNA 或 (和)组蛋白 ]),如慢性粒单核细胞白血病、骨髓增生异常综合征、急性髓性白血病、肾透明细胞癌和副神经节瘤等【35-39】。而TET蛋白催化的主动脱甲基作用,以及DNA甲基转移酶(DNA methyltransferase ,DNMT)耗竭或缺失导致的被动脱甲基作用,是肿瘤研究的重要方向【40,41】。
人体组织中表达 TET1、TET2、TET3共3种 TET蛋 白,TET蛋白可以将5-甲基胞嘧啶(5-methylcytosine,5mC)氧化为5-羟甲基胞嘧啶(5-hydroxymethylcytosine,5hmC)进而进一步氧化为5-甲酰基胞嘧啶(5-formylcytosine,5fC)、5-羧基胞嘧啶(5-carboxylcytosine,5caC),这些氧化产物不能被维持性DNMT识别,但在DNA链中可被胞嘧啶原型替换,因此降低DNA甲基化水平【42,43】。体内外研究均表明, TET蛋白活性增强可提高细胞 5hmC水平、降低细胞恶性程度【43,44】。组蛋白脱甲基酶(histone demethylases,HDMs)可移除组蛋白中精氨酸和赖氨酸上的甲基,该酶家族中的 Jumonji-C区组蛋白脱甲基酶(Jumonji C-domain-containing histone demethylases,HDMs)类似TET蛋白,作为Fe²+/α-KGDDS家族的一员催化脱甲基反应【45】。维生素C作为Fe²+/α-KGDDS的辅助因子能够增强或补偿现存TET蛋白的功能,而维生素C缺乏则会加速TET蛋白突变相关的肿瘤;同样,维生素C能够增强JHDM的脱甲基化作用,而JHDM功能受抑制时发生肿瘤的倾向增强【31,32,46】。体外试验表明,添加维生素C能够增强肿瘤细胞TET蛋白活性,使得细胞内5hmC水平升高、5mC水平降低,表现为细胞恶性程度降低、对抗癌药物敏感性增强【47,48】;在小鼠中,补充维生素 C能够增强TET蛋白活性、提高5hmC水平,延缓移植的基因易感的造血细胞发展为急性髓细胞白血病的进程【31】。
通过维生素C表观遗传学的调节作用杀伤肿瘤细胞是肿瘤治疗的一个研究方向,虽然目前的研究主要限于细胞实验和动物实验中维生素C对肿瘤细胞TET蛋白活性的影响、体细胞中维生素C通过调节JHDM活性对细胞分化的影响等方面,但维生素C治疗血液肿瘤的临床应用研究也已逐步展开。维生素C此作用机制的研究仍需更多的研究数据和证据支持。
3.维生素C调节HIF-1的活性
HIF-1是由受氧含量调节的HIF-1α和持续表达的HIF-1β构成的一种异二聚体转录因子,它可通 过上调细胞糖酵解、血管生成、细胞存活通路、红细胞生成和组织重塑等作用使得细胞适应缺氧和代谢应激状态;HIF-1在固体肿瘤缺氧的微环境中被激活,并被认为是肿瘤生长、转移、放化疗耐受的关键介质, 因此 HIF-1亦是肿瘤治疗的重要靶点【49-51】。HIF-1α的活性受 HIF羟化酶调节,该酶由3个脯氨酸羟化酶结构域蛋白(proline hydroxylase domain proteins,PHD)(即PHD1、PHD2、PHD3)和天冬氨酸羟化酶(即HIF抑制因子,factor-inhibiting HIF,FIH)构成,属于 Fe²+/α-KIDDs,但其对氧气分子的亲和力相对较低,导致缺氧环境下PHD和 FIH失活,而 HIF-1稳定并活化【52,53】。HIF羟化酶调节 HIF-1α的机制为:PHD可将HIF-1α的脯氨酸残基羟基化,脯氨酸羟化的HIF-1α被希佩尔-林道(von Hippel-Lindau,VHL) 肿瘤抑制蛋白结合,继而激活 E3-泛素连接酶,致使 HIF-1α被降解;而FIH可将HIF-1α的天冬酰胺残基羟化,这个羟化的基团抑制共激活蛋白p300与HIF复合物结合,从而抑制了HIF-1的转录活性并阻断了其下游通路【54】。
维生素C作为 Fe²+/α-KGDDs的辅助因子,理论上能增强 HIF羟化酶的功能。体外试验中,维生素C能增强细胞内HIF羟化酶活性、抑制HIF-1转录反应:维生素C干预甲状腺癌细胞能够诱导HIF-1α表达下降,并存在剂量反应关系;动物实验中,移植肺腺癌组织的维生素C缺乏小鼠模型经大剂量维生素C干预后,HIF-1α的表达和微血管密度均下降;而比较人体肿瘤标本(子宫内膜癌、肾细胞癌、结直肠癌)时发现,维生素C缺乏的肿瘤组织HIF-1活性最高【55-59】。通过维生素C调节HIF-1的活性, 进而控制或治疗肿瘤的研究结果仍有待证实,但关于维生素C对HIF-1的影响的结论是相对一致的。
综上所述,维生素C用于肿瘤治疗是有较充足的证据支持的。维生素C作为一种还原性必需营养素,发挥其正常生理作用之余,可通过诱导过氧化应激反应、调节表观遗传学表达、调节 HIF-1 的活性杀伤肿瘤细胞、降低肿瘤细胞恶性程度、增强肿瘤对治疗的敏感性。但关于维生素C抗肿瘤机制方面仍有许多关键问题亟待分析:
①剂量问题,维生素C通过诱导过氧化应激反应杀伤肿瘤细胞时需要超过生理浓度数十倍至数百倍的浓度水平,而生理浓度的维生素C即可发挥表观遗传学调节作用,故应用维生素C抗肿瘤时是否需要按照治疗目的选择剂量值得探讨;
②敏感性问题,大剂量维生素C可特异性杀伤肿瘤细胞而不损害正常细胞,而且仅部分肿瘤或肿瘤细胞系对维生素C干预敏感,造成这些差异的分子基础需要深入分析;
③多种机制假说的关联,维生素C作用于细胞时以上3种机制是否同时发生、相互之间有何影响、如何将维生素C的抗肿瘤作用最大化发挥等问题也应是维生素C抗肿瘤机制研究的研究方向。
关于维生素C抗肿瘤机制研究的深入开展,将为肿瘤治疗提供新的思路,为其他抗肿瘤治疗方法及药物提供新的靶点【60】。





转载自:石汉平医生

